However, Ernesto Estrada, a mathematician and expert in complex systems of the ARAID Foundation at the University of Zaragoza, decided to focus on finding targets within SARS-CoV-2 for new drugs to attack. From previous work, he knew that the main protease of the virus, an enzyme in charge of proteolytic processing of polyproteins, is an excellent target.
In the journal Chaos, from AIP Publishing, Estrada said when he and colleagues discovered a dramatic increase in the sensitivity of the main protease of SARS-CoV-2 to small disturbances, it made them suspect there is a role for inhibitors to play in killing the virus.
Inhibitors are organic molecules, drugs, or new chemical compounds that attach to a protease’s binding site to inhibit its work. A virus will die without a proteolytic enzyme working for it.
“I noticed that chemists had already found some potent inhibitors of the main protease of SARS-CoV-2, and that they’d resolved the structure of this protein via X-ray crystallography,” he said. “It was shocking to see that this protease is very similar to that of the SARS coronavirus, which produced the epidemics of 2003, SARS-CoV-1.”
When researchers superimposed both structures on top of each other, they matched almost perfectly.
“If you line up the amino acid sequences of both proteases, there are only 12 out of 306 residues that don’t coincide,” Estrada said. “Is there something hidden behind these apparent similarities between the two proteases? Can we learn something from them to improve the design of drugs against the virus?”
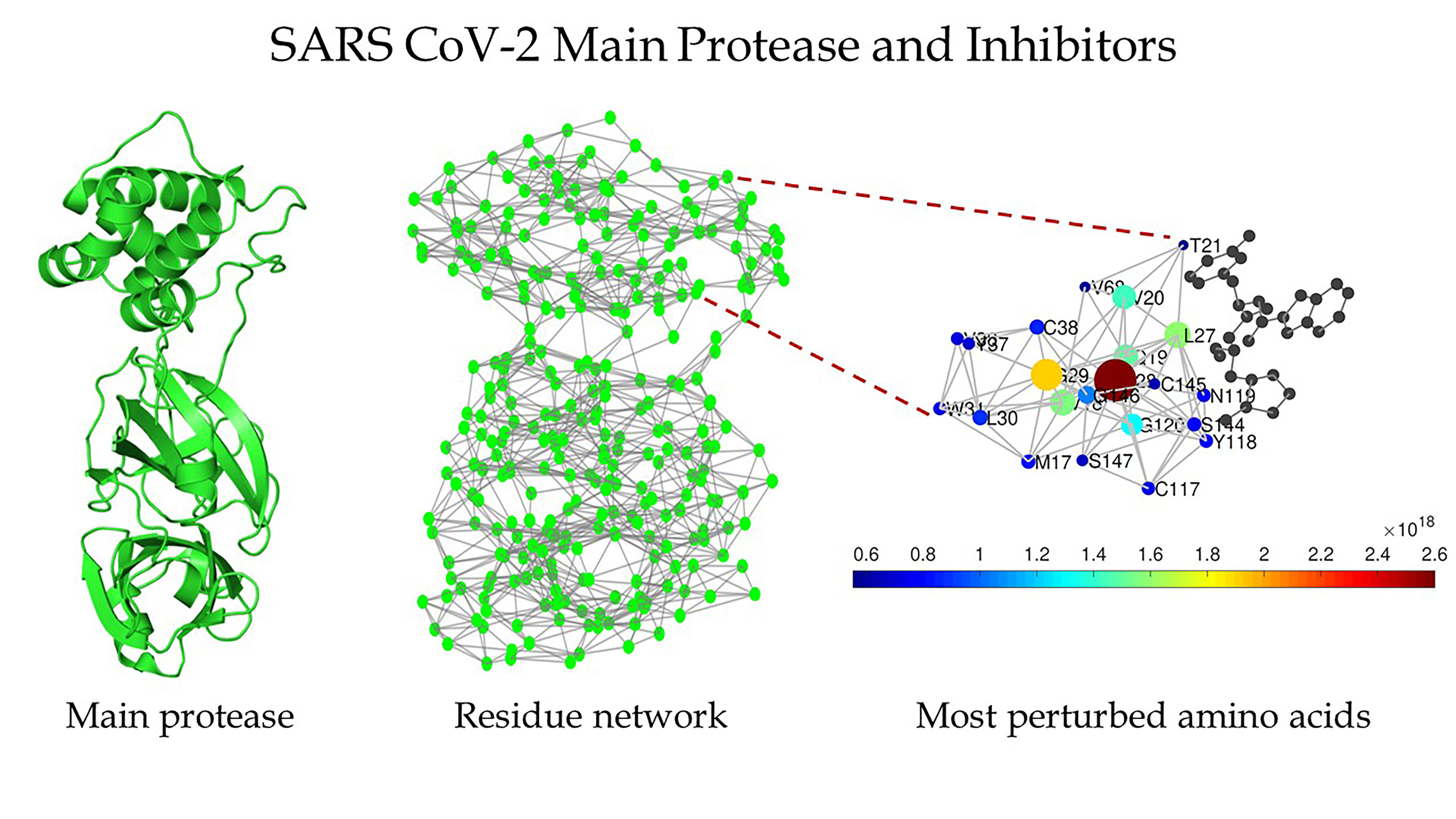
Estrada’s group has extensive experience in the analysis of networks — like social networks, the internet, or food chains between species within an environment — and decided to treat a protein as network.
“They’re called protein residue networks, where we represent every amino acid as a node, and the interaction between two amino acids is represented by a link between the two,” he explained.
They found several structures of the main protease of SARS CoV-1 and SARS CoV-2 that were clean, which means that they contain no mutations, ligands, or solvents within their structures. They transformed their structure into protein residue networks.
Estrada said most traditional network measures revealed both structures were very similar to each other, something his team already knew. “But a couple of years ago, we developed a more sophisticated mathematical measure that allows us to detect how far away a perturbation within a network can be propagated. That work was of a very theoretical, mathematical nature, but we had speculated that it could be useful for the study of proteins.”
So they put it to the test. It revealed that the protease of SARS-CoV-2 is 1,900% more sensitive to the long-range transmission of perturbations than the protease of SARS-CoV-1.
“This means that when a protein is perturbed, for instance by water within the intracellular environment, such perturbations are transmitted through a network of intraresidues that form the 3D structure of the protein,” Estrada said. “If such perturbation is produced around a given amino acid within the protease of SARS-CoV-1, it’s transmitted only through a close environment around that perturbed amino acid.”
But if this perturbation occurs to one amino acid within the protease of SARS-CoV-2, it is transmitted to almost the whole network — even to amino acids very far away.
“It’s remarkable, because it means that with tiny structural differences the protease of SARS-CoV-2 is much more effective within intraresidue communications,” Estrada said. “It should be much more effective in doing its job as a proteolytic enzyme of the virus. The devil did a nearly perfect job here, but he left the door open. This great sensibility of the SARS-CoV-2 protease to perturbations can be its Achilles’ heel in relation to inhibitors.”
The group’s approach can be used for massive screening protocols to identify potent inhibitors of SARS-CoV-2 main protease and, consequently, for the development of new drugs to kill it.
###
The article, “Topological analysis of SARS CoV-2 main protease,” is authored by Ernesto Estrada. It will appear in Chaos, June 9, 2020 (DOI: 10.1063/5.0013029). After that date, it can be accessed at https://aip.scitation.org/doi/10.1063/5.0013029.
ABOUT THE JOURNAL
Chaos is devoted to increasing the understanding of nonlinear phenomena in all areas of science and engineering and describing their manifestations in a manner comprehensible to researchers from a broad spectrum of disciplines. See https://aip.scitation.org/journal/cha.
###
Original post https://alertarticles.info

