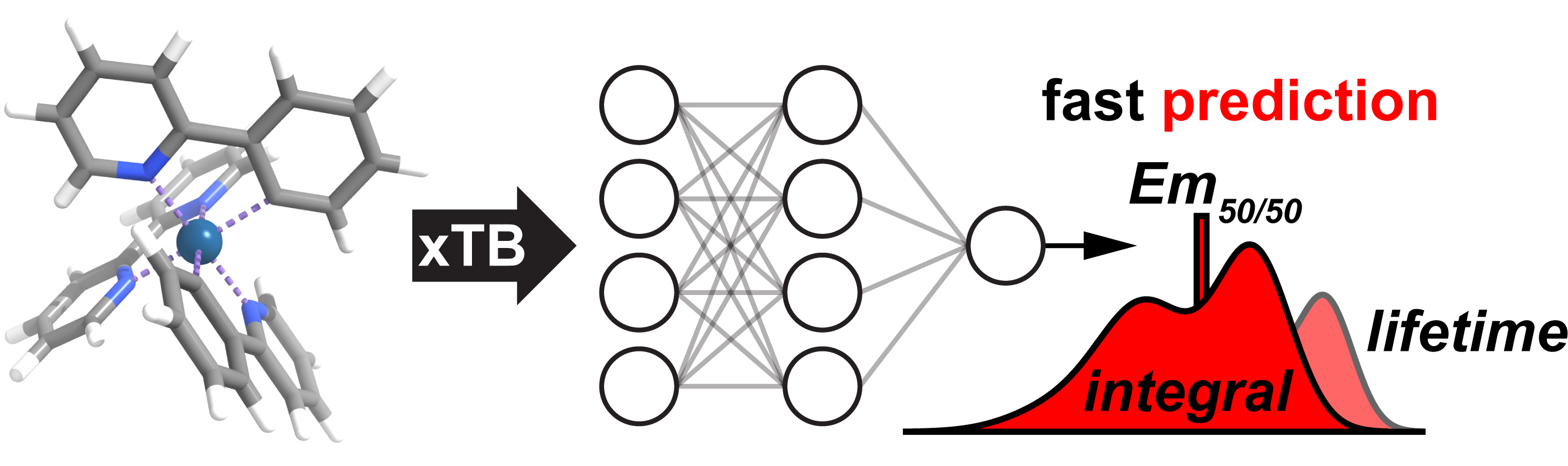
Kulik, an associate professor of chemical engineering, recently worked with Gianmarco Terrones, MIT chemical engineering graduate student, on simulations of high performance iridium phosphors – luminescent substances. Kulik and Terrones used Expanse to conduct the study which was recently published in Chemical Science.
The study, titled “Low-cost machine learning prediction of excited state properties of iridium-centered phosphors,” demonstrated the development of fast, accurate models that assess phosphor properties such as color and duration of light emission. The research represents one of the first applications of machine learning to the specific chemistry of iridium-centered complexes and revealed design rules for the synthesis of iridium phosphors with desired properties , such as emission lifetime.
What exactly are iridium phosphors?
Iridium phosphors are a type of chemical in which chemical building blocks called ligands are bonded to a central iridium atom. These chemicals are useful for a variety of applications such as organic light-emitting diodes (OLEDs) and photocatalysis. Choosing the best chemical building blocks to use for a phosphor is a challenging problem experimentally, since chemists are limited in the number of experiments they can run. To help with this, simulations on high-performance supercomputers such as Expanse can identify promising building blocks before any synthesis takes place.
“Our research focuses on the use of data-driven computer models (i.e., machine learning), which have a speed advantage over the usual ab initio first principles computer modeling approach – the data-driven models can be trained directly on experimental data as well, and can thus bypass certain accuracy limitations of ab initio models,” Terrones said. “These data-driven models can be used to identify good phosphors and bad phosphors, and answer questions like, does this type of ligand make a phosphor brighter or dimmer (leading to design rules).”
According to Kulick and Terrones, thanks to the Expanse calculations, other chemists will have an easier time synthesizing high-performing phosphors by using the developed artificial neural networks (ANNs), or the data-driven computer models, to quickly screen thousands of complexes and identify promising ones. In other words, they can now see what an ANN model thinks of a proposed new phosphor, and either proceed with synthesis or not – depending on the model verdict.
“Our work allows fellow chemists to efficiently search an infinite chemical design space by only considering phosphors that are likely to be high-performing,” Terrones said. “As chemists go on to synthesize new phosphors, computational researchers like us can use the new phosphors as examples to feed to computer models, which then learn more and become capable of making better predictions. As a result, there is a feedback cycle between model and experiment that helps both advance further than either could alone.”
How did using Expanse make a difference?
Data-driven models on Expanse, like those created by Kulik and Terrones, have the power to accelerate chemical discovery, and the researchers say that their application to iridium phosphors will lead to faster discovery of efficient photocatalysts for green chemistry and optimal iridium phosphors for efficient, vibrant OLED technology and bioimaging.
“Access to Expanse allowed for time-dependent density functional theory (TDDFT) calculations of dozens of iridium phosphors and enabled the benchmarking of data-driven computer models with TDDFT, the latter of which is commonly used to study iridium phosphors,” Terrones said. “Expanse was also used for the training of ANNs. The application of our models to thousands of hypothetical iridium complexes derived from the Cambridge Structural Database in a matter of seconds was very satisfying as it highlighted the usefulness of the models for chemical discovery.”
The lab’s next step is to apply the developed models to an active learning workflow in order to identify additional promising phosphors. In this approach, the goal is to attain edge-of-distribution combinations of emission energy and lifetime by retraining the models on ab initio data of phosphors identified as optimal by their Expanse models.
Additional scientists working on the study were MIT researchers Chenru Duan and Aditya Nandy. The Office of Naval Research (grant no. N00014-18-1-2434 and grant no. N00014-20-1-2150) provided primary support for this work. Support for machine learning feature development was provided by DARPA (grant no. D18AP00039). Computational work on SDSC resources was supported by National Science Foundation (NSF) Extreme Science and Engineering Discovery Environment (grant no. ACI-1548562). Additional support was received from the Alfred P. Sloan Foundation (grant no. G-2020-14067) and the NSF Graduate Research Fellowship Program (grant no. 1122374).

