The number of people infected with the virus varies around the world. Less than 10% of people in the U.S. are infected with KSHV, compared to 50% of the population in some parts of Africa. Not everyone with KSHV will develop Kaposi sarcoma. Those who do, generally have a weakened immune system due to HIV infection, organ transplant, being older or other factors.
The introduction of antiretrovirals to control HIV significantly reduced AIDS-related Kaposi sarcoma prevalence in Western countries; however, in sub-Saharan Africa, the disease continues to have a poor prognosis.
What keeps the Kaposi’s sarcoma-associated herpesvirus dormant?
When the virus enters a human cell, it causes a hidden infection in the nucleus. During this stage, the virus is latching onto parts of the cell’s chromosomes and not producing viral offspring.
A study published in Cell Reports looked at KSHV’s latent-lytic switch, a process in which the virus exits its dormancy state to replicate in the host cell. This replication phase, called the lytic cycle, ends with the disintegration of the cell and the release of the viruses, infecting neighboring cells.
“The virus likes to stay silent as long as possible to avoid being detected by the body’s immune system,” said Yoshihiro Izumiya, the study’s senior author. Izumiya is a professor at the Department of Dermatology and director of the Viral and Pathogens Associated Malignancies Program at UC Davis Comprehensive Cancer Center.
The researchers wanted to uncover the mechanisms behind this latent-lytic switch and the role the host cell environment played in this process.
“Where the virus latches onto the host cell, how it manages to stay dormant, and what triggers its activation were very exciting and important puzzles to solve,” Izumiya said.
Finding the preferred ecosystem for the virus to stay dormant
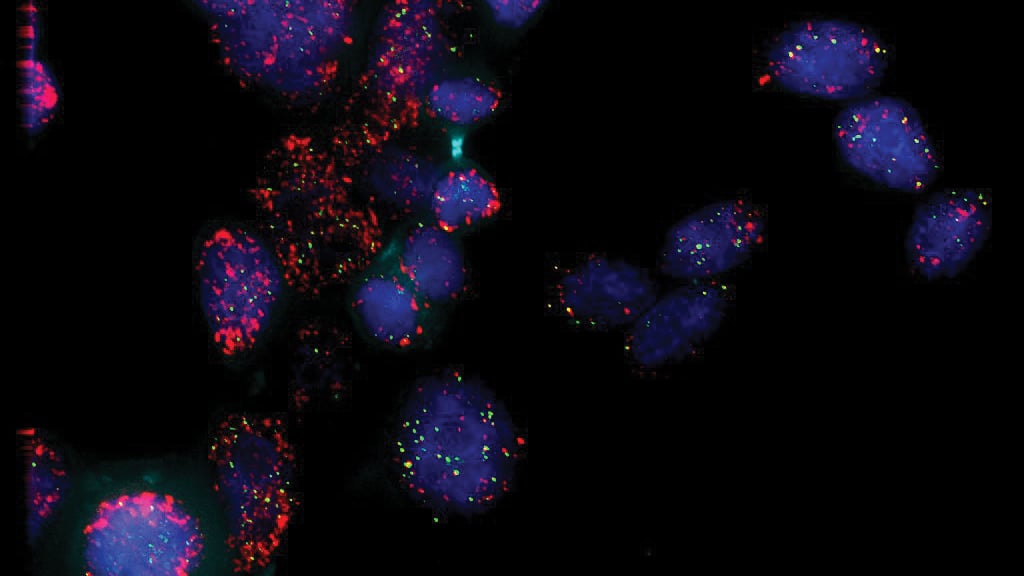
The study identified where the virus genome could be found on the host genome.
Izumiya and his team used Capture Hi-C and DNA FISH methods to profile and analyze chromosomal interactions on three cancer cell lines naturally infected with KSHV. They located the virus’s preferred docking sites inside the host chromosomes. The binding patterns, similar among the three cancer cell lines, showed a nuclear ecosystem that can attract and help keep the virus in its silent form.
The team also found that CHD4 (chromodomain helicase DNA binding protein 4) binds to the virus’s genomic elements. CHD4, a protein in the host cell’s chromosomes, suppresses the work of the gene responsible for viral replication. The study showed that CHD4 is a key regulator of the KSHV latency-lytic switch.
“The location where the virus genome attaches to the host chromosome is not random,” said Ashish Kumar, a postdoctoral researcher in Izumiya Lab and the paper’s first author. “Without having enriched CHD4 protein, the virus starts to replicate, kicking in a cell destructive mode. For the virus to select CHD4 among many other host proteins, CHD4 must play a unique and important role in host cells.”
Evolution shapes strategic viral protein binding to host
The study of viruses, known as virology, can help identify cellular proteins essential for cell homeostasis. Over millions of years, the virus’s genome developed to encode or assemble a small number of very efficient proteins. These proteins strategically connect to host cell proteins to keep viral chromatin dormant and impact the host cell’s tumor suppression function.
“We used virology as an entry point to shed light on the function of CHD4 in gene regulation in general. During virus-host co-evolution, KSHV cleverly learned to hijack host proteins that can help keep the gene responsible for viral replication dormant.”
The researchers discovered a viral protein that impacts the CHD4 function. They pointed to the potential of using viral protein sequence as a starting point to create inhibitors regulating CHD4 function. As CHD4 is critical for cancer cell growth in many different types of cancers, they hope their work will inform cancer therapy development by utilizing this virus-host interaction.
The study is a collaboration among UC Davis researchers from the Genome Center, UC Davis Comprehensive Cancer Center and the Departments of Dermatology, Biochemistry and Molecular Medicine, and Pathology and Laboratory Medicine. It is also in partnership with researchers at the HIV Dynamic and Replication Program at the National Cancer Institute (NCI) and the Lifescience Division of Lifematics in Japan.