
CHAPEL HILL, NC – October 21, 2020 – Babies born with a faulty maternal copy of the UBE3A gene will develop Angelman syndrome, a severe neurodevelopmental disorder with no cure and limited treatments. Now, for the first time, scientists at the UNC School of Medicine show that gene editing and gene therapy techniques can be used to restore UBE3A in human neuron cultures and treat deficits in an animal model of Angelman syndrome.
This work, published in Nature and led by senior author Mark Zylka, PhD, Director of the UNC Neuroscience Center and W.R. Kenan, Jr. Distinguished Professor of Cell Biology and Physiology, lays important groundwork for a long-lasting treatment or cure for this debilitating disease, as well as a therapeutic path forward for other single-gene disorders.
“Our study shows how multiple symptoms associated with Angelman syndrome could be treated with a CRISPR-Cas9 gene therapy,” Zylka said. “And we are now pursuing this with help of clinicians at UNC-Chapel Hill.”
Angelman syndrome is caused by a deletion or mutation of the maternal copy of the gene that encodes the ubiquitin protein ligase E3A (UBE3A). The paternal copy of UBE3A is typically silenced in neurons, so the loss of maternal UBE3A results in a complete absence of the UBE3A enzyme in most areas of the brain. That’s crucial because the enzyme targets proteins for degradation, a process that maintains normal function of brain cells. When that process goes awry, the result is Angelman syndrome, a brain disorder with symptoms that include severe intellectual and developmental disabilities, seizures, and problems with speech, balance, movement, and sleep.
“Turning on the paternal copy of UBE3A is an attractive therapeutic strategy because it could reverse the underlying molecular deficiency of the disease,” Zylka said. However, the paternal gene is silenced by a long strand of RNA, produced in the antisense orientation to UBE3A, which blocks production of the enzyme from the paternal copy of the gene.
Members of the Zylka lab, including postdoctoral fellows Justin Wolter, PhD, and Giulia Fragola, PhD, set out to devise a way to use CRISPR-Cas9 to restore the UBE3A enzyme to normal levels by disrupting the antisense RNA. Preliminary data in cell cultures were promising, and Zylka received grants from the NIH, the Angelman Syndrome Foundation, and the Simons Foundation to test their findings in human neurons and in a mouse model of the disease.
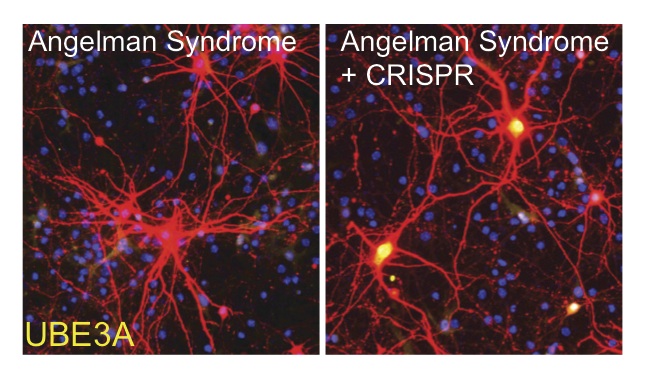
In the Nature paper, co-first authors Wolter and Hanqian Mao, PhD, a postdoc in the Zylka lab, and UNC colleagues describe using an adeno-associated virus (AAV) gene therapy to deliver the Cas9 protein throughout the brain of embryonic mice that model Angelman syndrome. Because UBE3A is essential for normal brain development, early treatment is crucial. The researchers found that embryonic and early postnatal treatment rescued physical and behavioral phenotypes that model core deficits found in Angelman syndrome patients. Remarkably, a single neonatal injection of AAV unsilenced paternal Ube3a for at least 17 months, and the data suggest this effect is likely to be permanent. The researchers also demonstrated that this approach was effective in human neurons in culture.
“We were blown away when we got these results,” Zylka said. “No other treatments currently being pursued for Angelman syndrome last this long, nor do they treat as many symptoms. I am confident others will eventually recognize the advantages of detecting the mutation that causes Angelman syndrome prenatally and treating shortly thereafter.”
Wolter added, “The results of treating early were very promising. Since we learned we could reduce the severity of Angelman syndrome in mice, we are now focused on refining our approach in ways that will be suitable for use in humans.”
While working to translate this research into the clinic, the Zylka lab will collaborate with researchers at the Carolina Institute for Developmental Disabilities (CIDD) to identify symptoms in babies that have the genetic mutation that causes Angelman syndrome.
Zylka’s lab is working with CIDD researchers led by CIDD director Joseph Piven, MD, to use brain imaging and behavior observations to identify symptoms associated with Angelman syndrome in infants. Anecdotal reports suggest these infants have difficulty feeding and reduced muscle tone, but these and other early symptoms have not been rigorously characterized to date.
“The idea is to use genetic tests to identify babies that are likely to develop Angelman syndrome, treat prenatally or around the time of birth, and then use these early symptoms as endpoints to evaluate efficacy in a clinical trial,” Zylka said. “Our data and that of other groups clearly indicate that prenatal treatment has the potential to prevent Angelman syndrome from fully developing.”
As part of the Nature study, the researchers also found that the gene therapy vector blocked the antisense RNA by integrating into the genome at sites cut by CRISPR-Cas9. This so-called “gene trap” could be exploited to disrupt other long non-coding RNAs and genes.
Zylka added, “We are incredibly excited to keep this work moving forward with the hope of helping children and families overcome this debilitating condition. Support from the NIH, the Simons Foundation, and the Angelman Syndrome Foundation was essential for moving this work forward.”
Along with Zylka, Wolter, and Mao, co-authors of the Nature paper are Giulia Fragola, PhD, postdoc in the Zylka lab at the time of this research; Jeremy Simon, PhD, research associate professor; James Krantz, Zylka lab research associate; Hannah Bazick, Zylka lab graduate student; Baris Oztemiz, Zylka lab research technician; and Jason Stein, PhD, assistant professor of genetics and member of the UNC Neuroscience Center; all at UNC-Chapel Hill.
This research was funded by grants from the National Institutes of Health, the Simons Foundation, the Angelman Syndrome Foundation, the Eshelman Institute for Innovation, and the Pfizer-NCBiotech Distinguished Postdoctoral Fellowship in Gene Therapy.
Media contact: Mark Derewicz, 919-923-0959

